



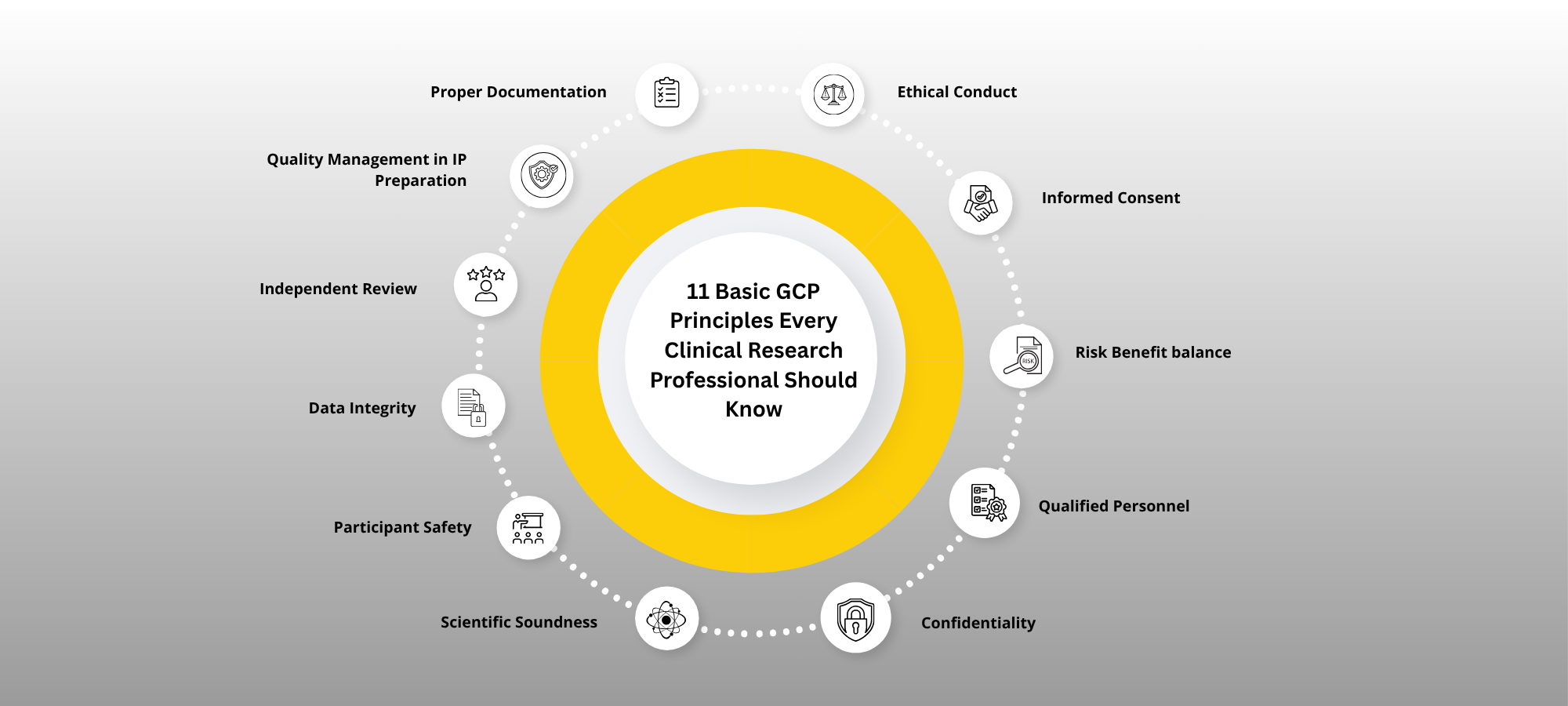
The International Council for Harmonisation (ICH) has long guided this process through its Good Clinical Practice (GCP) guidelines. The latest revision, ICH-GCP E6(R3), introduces 11 core principles that reflect evolving technologies, trial designs, and global expectations.
Let’s explore each of these principles in detail:
1) Ethical Conduct Based on the Declaration of Helsinki
Clinical trials must be conducted in accordance with ethical principles derived from the Declaration of Helsinki, prioritizing the rights, safety, and well-being of trial participants above all other considerations
2) Informed consent
Participation in clinical research must be voluntary, and based on a process of adequate and comprehensible informed consent. Participants or their legally authorized representatives must be fully informed of the nature, risks, and potential benefits of the study.
3) Independent review by an IRB/IEC.
All clinical trials must undergo review and approval by an Independent Ethics Committee (IEC) or Institutional Review Board (IRB). This ensures that the study protocol adheres to ethical standards and safeguards participant welfare.
4) Scientifically sound and Fit-for-Purpose Design
Clinical trials must be designed to be scientifically robust and appropriate for their intended objectives. This includes the application of current scientific knowledge and methodologies to ensure meaningful and reliable outcomes.
5) Qualified individuals.
Individuals involved in the design, conduct, monitoring, and evaluation of clinical trials must possess the appropriate qualifications, training, and experience to perform their respective roles competently.
6) Quality should be built into the Trial design and conduct.
Clinical trial quality means being fit for purpose, protecting participants and ensuring reliable results. Identifying critical-to-quality factors early helps meet trial goals. Strong strategies must prevent and manage serious noncompliance with GCP and regulations
7) Proportionality of Risk and Complexity.
Trial procedures and oversight should be proportional to the risks and complexity of the study by focusing on participant safety and data reliability. Risks beyond routine care, especially with investigational products, must be considered. Processes should be simple, feasible, and support key objectives without burdening participants or investigators.
8) Clinical trials should be described in a clear, concise, scientifically sound and operationally feasible protocol.
Clear, practical trial protocols with well-defined goals and supporting plans ensure participant safety and reliable results.
9) Clinical trials should generate reliable results.
Trial data must be fit for purpose accurate, reliable, and sufficient for decision-making. Systems for data capture and analysis should be risk-proportionate and ensure integrity. Computerized tools must be validated and designed to protect data. Efficient record management is essential for traceability and privacy. Sponsors must retain essential records and ensure transparency through public trial registration and result sharing.
10) Roles and responsibilities in clinical trials should be clear and documented appropriately.
In clinical trials, roles and responsibilities must be clearly defined and documented. While sponsors can transfer and investigators can delegate tasks, they retain overall responsibility for their respective activities. All agreements should specify who is responsible for what, especially when involving third-party service providers. Regardless of delegation, sponsors and investigators must maintain proper oversight to ensure trial quality and data integrity.
11) Products must follow GMP and be managed per product specifications and protocol.
Ensure GMP-compliant manufacturing, maintain product quality, follow protocol use, label per regulations, and manage product logistics securely.